
テクニカルアドバイス
支援センターでは、研究・技術支援やDNAシーケンス・DNAマイクロアレイの受託を行っています。基本的なことだけれどなかなかうまくいかない、という利用者の方からのご質問に対して、支援センターで行っている方法のいくつかを以下に挙げています。
![]() DNAシーケンス用のプラスミド抽出の方法
DNAシーケンス用のプラスミド抽出の方法
![]() DNAシーケンス反応・精製プロトコール
DNAシーケンス反応・精製プロトコール
![]() Total RNA抽出の注意点
Total RNA抽出の注意点
![]() リアルタイムPCRの予備実験
リアルタイムPCRの予備実験
各社より市販されているプラスミド精製キットを用いれば確実ですが、以下の方法で精製したプラスミドを用いても各ステップを丁寧に行えば500b~700bまで読めています(クラボウ自動DNA抽出装置と同程度の精製)。
1) LB培地2ml(Lowコピーのプラスミドの場合5~10ml)で大腸菌を終夜培養。
2) 1.5mlをエッペンドルフチューブ(10ml遠心用チューブ)へ移し、集菌(10,000 rpm、4℃、 5分) 、 上清を完全に除去する。氷中へ置く。
3)
大腸菌(沈殿)にTE/RNAse バッファー200μl(1ml)を加えてボルテックスで懸濁。
4) Sol 2, 200μl(1ml)を添加し、転倒混和を10回行い室温で5分静置(Total5分、変性)。
5) 更に、Sol 3, 200μl(1ml)を添加し転倒混和を10回行い、氷中で5分以上静置(中和)。
6) 15,000 rpm、4℃、 5分間遠心し、上清を新しい1.5 mlエッペンドルフチューブに移す。
(白い沈殿物が入らないように注意する)
7) 移した上清にフェノール・クロロホルム500μlを加え、転倒混和3分。
8) 15,000 rpm、4℃、5分間遠心し、下層のフェノール層を除去する(少し残っても構わない)。
9) そこにクロロホルム500μlを加え、転倒混和3分。
10) 15,000 rpm、4℃、5分間遠心し、上清を新しい1.5 mlエッペンドルフチューブへ移す(中間層を取らないように注意)。
11) 等量のイソプロパノールを加え十分に撹拌後、室温30分静置後、15,000 rpm、4℃、20分間遠心し、上清を廃棄する。
12) 沈殿のDNAに500μlの70 % エタノールを加え、15,000rpm、4℃、5分間遠心し、上清をできるだけ除去し、風乾2分。乾燥しすぎないように注意する。
13) 沈殿のDNAに20μlのdH2O(TE)を入れ、氷中2分静置後タッピングで溶解する
14) DNAを1μl*1電気泳動し、DNAマーカー(サイズだけではなく、濃度も分かるマーカー)のバンドと比較しておおよその濃度を算出する*2。吸光度(OD260)の測定もする。
15) シーケンス用プロトコールに従い、シーケンス反応を行う。
「注意点」
*1大腸菌の中でコピー数の少ない(pBR322系)プラスミドの場合は2μl電気泳動して下さい。
*2吸光度OD260は、染色体の残骸やRNAも入ってきますので過剰に加味されます。きれいなDNAだとアガロースゲル電気泳動の値と吸光度計の結果はほぼ同じです。
| 試薬リスト |
| TE: 10 mM Tris, 1 mM EDTA-Na2, pH 8.0 TE/RNAse: TE containing 2 U/ml RNAse Sol 2: 0.2 M NaOH, 1 % SDS Sol 3: 3M KOAc, pH 4.8 フェノール・クロロホルム:TEで飽和させたフェノール:クロロホルム=1:1でmix クロロホルム |
 DNAシーケンス反応・精製プロトコール
DNAシーケンス反応・精製プロトコール
(Big Dye Terminator サイクルシーケンシングプロトコール)
使用キット:Applied Biosystems Big Dye Terminator v3.1 Cycle Sequencing Kit
シーケンスを行いたいサンプルとプライマー及びプレミックスを以下の組成で調製します。
| ・バッファー(Big Dye Sequencing Buffer) | 3.5μl | |
| ・プライマー | 3.2 pmol | |
| ・テンプレートDNA ( dsDNA or PCR 産物 ) | Xng*1 | |
| ・プレミックス (BigDye Terminator v3.1Cycle Sequencing Kit Code No. 4303152) |
1.0μl | |
| 滅菌水にて 計20μlにメスアップ | ||
サンプルを泡立てないようにゆっくりと混和させ、以下の条件でサーマルサイクラーにセットし、PCR 反応(*2、*3)を行います。
96℃ 1分*4
↓
96℃ 10秒┐
50℃ 10秒├25 サイクル
60℃ 4分┘
↓
4℃
シーケンス反応後、サンプルの精製を行い、反応で取り込まれなかった色素を取り除く必要があります。(下記参照)
「注意点」
*1 サンプルのサイズに応じて使用量を決めます。プラスミドDNA6,000bpで100ng(アガロース電気泳動の濃度で)使用します。
*2 このPCR 温度条件は、ABI GeneAmp PCR システム モデル9600,9700,2400,2700を用いた場合の設定値です。各社サーマルサイクラーがありますが、各社でスロープの違いがありますので温度条件、反応時間等はご検討ください。またアニーリング温度は50℃となっていますが、シーケンスがうまく読めない時や2重構造があって途中でシーケンス反応ががくっと止まってしまう時は、Tm値から判断して変更してみて下さい。
*3 ミネラルオイルを使用する機種はサイクルが終了したらミネラルオイルをパラフィルムなどで除いてください。
*4 ヒートブロックが90℃以上になってからチューブを差し込みます。
サンプルの精製方法
以下にあげるいずれかの方法でサンプルを精製してください。エタノール沈殿を用いた方法では一部フリーの蛍光Terminator が残ることがあります。
(I)スピンカラム(Centri-Sep: ABI P/N 401762, 401763) を利用した方法
1) カラム内へ滅菌水800μl を加える。
2) 2時間以上静置し、乾燥したゲルを十分に水和させる。その後、 ボルテックスで撹拌する。
3) カラムに気泡がないこと(気泡がある場合は、カラムを軽くたたくことにより除去する)を確認する。
4) 上のキャップ、下のストッパーの順に外す(キャップは必ず上から外します。下を先に外してしまうとゲル内に気泡が入ってしまいます)。
5) ウォッシャーチューブをセットする。
6) カラム内の滅菌水をゲル表面まで自然落下させる。
7) 捕集した滅菌水を捨てる。
8) 再びウォッシャーチューブをセットする。
9) 730 x g (2,500 rpm )で、2分間遠心する(遠心機にセットする方向はいつも 同じにし、325 x g-730 x g で遠心します。2分間以上遠心しないようにします。また、735
x g 以上で遠心しないようにします。)。
10) サンプルチューブをセットする。
11) 反応液20μlをカラムの中央にアプライするチューブの壁などに触れないように注意します。反応液が壁に付いた場合フリーの蛍光Terminator
が残る原因になります)。
12) 730 x g (2,500 rpm )、2分間遠心する。
13) サンプルチューブに回収されたサンプルをSpeed vac などを用いて乾燥させる。
(II)エタノール沈殿を利用した方法
1) 20μl反応液に2μlの125mM EDTAを各 ウェルに添加する(加える時には、サンプルに直接混ざるように入れる事)。
2)2μlの3Mの酢酸ナトリウムを各サンプルに添加する(加える時には、サンプルに直接混ざるように入れる事)。
3)50μl~60μlの100%のエタノール(室温*1)を添加する(2.5vol)。
4)5~6回転倒混和して十分に撹拌する。
5)室温で15分間インキュベートする。
6)15000rpmで20分室温*1で遠心する。
・沈殿は見えないので、キャップの根元に沈殿がくるように遠心を行う。
・p200のピペットマンで、根元とは逆の方から上精をすばやく取り除く。
7)その沈殿に70μlのフレッシュな70%エタノール(室温*1)を加える。
8)15000rpmでは5分室温*1で遠心する。
9)p200のピペットマンで、根元とは逆の方から上清を丁寧に取り除く*2
10)上清を取り除いた後、37℃で2分乾燥する。
(アルコールのにおいがしない事を確認する*3)
11)シーケンサーにかける時まで遮光して-20℃で保存。
「注意点」
*1エタノールと遠心機は室温で使用してください。冷えているとフリーの蛍光Terminatorが落ちてきて、塩基配列の特定の箇所が読みにくくなります。
*2上清を残さないように注意深く吸い取って下さい。
*3アルコールは、シーケンサーの電気泳動を阻害します。タッピングして少しも水分(アルコールのにおいがしないか)がないことを確認して下さい。

リアルタイムPCRがなかなかうまくいかない、という方の中には予備実験をされてない方が多くいらっしゃいます。実際に大量のサンプルをかける前に、下記のことをご確認ください。
| 1. Total RNAの品質チェック |
| 2. Standard Curve解析 |
| ☆ 定量可能範囲の決定 |
| ☆ 増幅効率と再現性のチェック |
| ☆ No RTのチェック |
| 以降のステップはSYBRGreenの実験系の方 |
| ☆ NTC(No template control)のチェック |
| 3. Dissociation Curve解析 |
1.
Total RNAの品質チェック・・・上記「Total RNA抽出の注意点」をご覧下さい。
元々intactなRNAと壊れているRNAで発現の差をみようとしても発現の差があるのか、壊れた結果の差なのか分かりません。毎回でなくて良いですので、初めてその実験系をされる時は、そのTotal RNA抽出の手技が大丈夫か確認されるために、Total RNAの品質チェックを是非されて下さい。電気泳動では、すごく壊れているRNAはバンドのサイズが違うところにたくさんでてきて分かりますが、そこまででないRNAの壊れ具合は判別することができません。

2. Standard Curve解析
プライマーごとに準備します。
・ cDNA(Total RNA換算で)50ng 1本
・ NTC(No template control) 1本
・ No RT*1 1本
*1 cDNAの元のRNAを逆転写する時、
Reverse transcriptase(RT)を加えず、代わりに水を入れて他は全く同じように反応を行ったサンプルです。ゲノムが 増幅されていないかの確認のために行います。

① 上記リアルタイムPCRでかけたPCR産物を1部アガロースゲル電気泳動をし、非特異的なバンドがないかプライマーダイマーがないかチェック
② またこのPCR産物を10,000分の1希釈し*2、それを1として10倍希釈ずつ(トリプリケイト)したものをつくる。
③ ②に試薬を入れて、Standard Curveをかく。
*2PCRかけているのですごく濃いことと、SYBRGreenやTaqの蛍光色素を希釈するためです。
*PCR産物の希釈系列を作成中に水・マスターミックス・プライマーなどにコンタミを起こしやすいので、希釈系列の作成はそれらがない場所で行う、またコンタミ防止用のUNG仕様のマスターミックスを利用するなどして下さい。
③
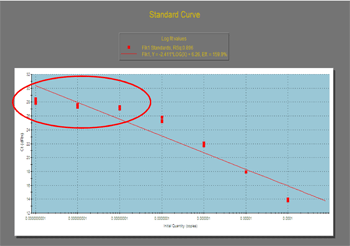
赤く囲んでいるところは、Standard Curveに傾きがないので、定量性がありません。ここの部分を削除したものが、下表になります。
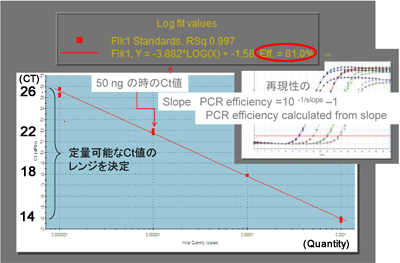
☆ 定量可能範囲の決定
cDNAを50ng使用した時のCT値は22でした。
左表より、定量可能なCt値のレンジは14ct~26ctの間であることが分ります。
| CT値 | 14 | 18 | 22 | 26 | |
| cDNA量(ng) | 5512 | 525 | 50 | 4.76 | |
Efficiencyは81%です。(下図1 )よりct値1サイクルの差は初期濃度が1.8倍の差となります。 |
|||||
本番で使用するcDNAのおおよその目安になります。プライマーごとに検討してますので、サンプルの中には微量しか発現しないものもあると思います。ケースbyケースでお考え下さい
。

☆ 増幅効率と再現性のチェック
PCRの増幅効率(Efficiency)を確認します。100%以下で安定した増幅効率(再度3回繰り返し実験を行って同様の結果が出る)であれば以降の実験はΔΔCt法で行うことができます。
← ΔΔCt法の『1Ctの差が初期濃度 2倍の差』というのは増幅効率が100%で計算されています。実際の増幅効率を100%のところに入れてExcelで計算して下さい。
上記の計算ができるExcelファイルを支援センターで準備していますので必要な方は声かけて下さい。
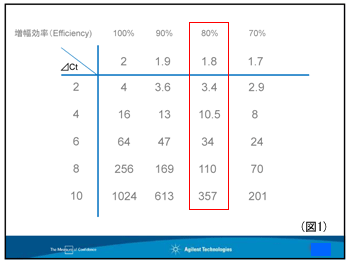
←増幅効率が違うとこんなにも初期濃度の差は変わってきます!
☆ No RT(逆転写反応なしのサンプル)のチェック
No RTでシグナルが出た場合、ゲノムのコンタミの可能性があります。unknownのCt値よりNo-RTのシグナルによるCt値が6.6Ct以上大きければ1%以下(PCRの増幅効率が100%の場合)の影響なので無視できる範囲内です。unknownとNo-RTのCt値の差が近い時はプライマーのデザインを再検討された方が良いです。
・・・ DNase処理について ・・・
エタ沈でサンプルをロスする可能性があります。どうしてもno-RTで増幅が出てしまう場合は、DNase処理するよりはプライマーデザインの変更がファーストチョイスで、どうしようもない場合にDNase処理をされて下さい。その場合には、ハウスキーピング遺伝子についてもDNase処理したものでやり直して下さい。。
以降のステップはSYBRGreenの実験系の方
☆ NTC(No template control)のチェック
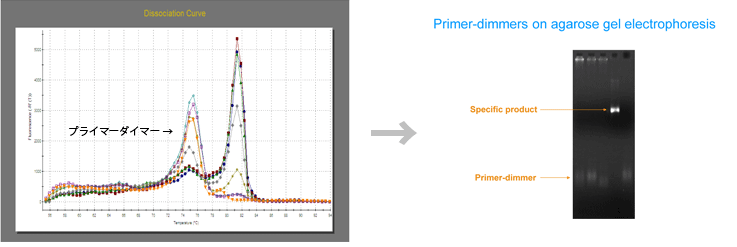
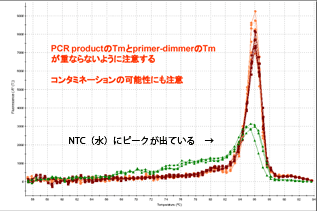 NTCでシグナルが出た場合、コンタミとプライマーダイマーの2種類の可能性があります。
Dissociation Curveやアガロースゲル電気泳動でどちらの原因かを考えます。
NTCでシグナルが出た場合、コンタミとプライマーダイマーの2種類の可能性があります。
Dissociation Curveやアガロースゲル電気泳動でどちらの原因かを考えます。
・ プライマーダイマーの場合・・・プライマーの再検討
・ コンタミが疑われる場合・・・試薬・primer・水など新しくしてみてコンタミの除去を検討。
3. Dissociation Curve解析
SYBRGreenの時は必須です。SYBRGreenは2本鎖DNAにインターカレートするので、プライマーダイマーや非特異的PCR産物も蛍光を発します。
GC含量が多いものや、プライマーサイズが長いものはTm値が高くなります。またcDNA少なかったり、発現量少なかったりすると、蛍光量のピークも低いです。
ピークが複数本出現していたり、何かおかしい現象のときにはプライマーの再検討など必要になってきます。
* 上記「リアルタイムPCRの予備実験」はアジレントテクノロジー株式会社ライフサイエンス部門 吉田 悟 氏よりアドバイスいただきました。
Copyright(C)The Research Support Center, Research Center for Human Disease Modeling,
Kyushu University Graduate School of Medical Sciences. All Rights Reserved.